Wall potential (MD)#
Overview#
Questions#
How do I apply interactions between wall geometries and particles in molecular dynamics simulations?
Objectives#
Demonstrate the use of a wall potential class.
Boilerplate code#
The render function in the next (hidden) cell will render the system state using fresnel. Find the source in the hoomd-examples repository.
Place particles in the center of a large simulation box:
[3]:
import itertools
import gsd.hoomd
import hoomd
import numpy
L = 20
m = 10
N = m**2
x = numpy.linspace(start=-m / 2, stop=m / 2, endpoint=False, num=m) + 1 / 2
position_2d = numpy.array(list(itertools.product(x, repeat=2)))
frame = gsd.hoomd.Frame()
frame.particles.N = N
frame.particles.position = numpy.stack(
(position_2d[:, 0], position_2d[:, 1], numpy.zeros(N)), axis=-1
)
frame.particles.types = ['mobile']
frame.configuration.box = [L, L, 0, 0, 0, 0]
with gsd.hoomd.open(name='initial_state.gsd', mode='x') as f:
f.append(frame)
Prepare a molecular dynamics simulation:
[4]:
simulation = hoomd.Simulation(device=hoomd.device.CPU(), seed=1)
simulation.create_state_from_gsd(filename='initial_state.gsd')
langevin = hoomd.md.methods.Langevin(kT=1.0, filter=hoomd.filter.All())
simulation.operations.integrator = hoomd.md.Integrator(dt=0.001, methods=[langevin])
Adding forces between particles and wall geometries#
In a molecular dynamics simulation, particles move in response to applied forces. Typically, this includes a pair forces between pairs of particles:
[5]:
pair_lj = hoomd.md.pair.LJ(nlist=hoomd.md.nlist.Cell(buffer=0.4), default_r_cut=2.5)
pair_lj.params[('mobile', 'mobile')] = dict(epsilon=1, sigma=1)
simulation.operations.integrator.forces.append(pair_lj)
This example gives the pair interaction a range of r_cut = 2.5. The simulation box is always periodic. To model an isolated system, place the wall geometries in from the edge of the box so that the particles do not interact across the periodic boundary conditions.
[6]:
top = hoomd.wall.Plane(origin=(0, 7, 0), normal=(0, -1, 0))
bottom = hoomd.wall.Plane(origin=(0, -7, 0), normal=(0, 1, 0))
left = hoomd.wall.Plane(origin=(-7, 0, 0), normal=(1, 0, 0))
right = hoomd.wall.Plane(origin=(7, 0, 0), normal=(-1, 0, 0))
Apply forces between the particles and the wall geometries using a hoomd.md.external.wall force:
[7]:
wall_lj = hoomd.md.external.wall.LJ(walls=[top, bottom, left, right])
The force due to a wall on a particle follows the form of a pair potential as a function of the distance between the center of the particle and the wall’s surface. Set the relevant pair potential parameters:
[8]:
wall_lj.params['mobile'] = dict(sigma=1.0, epsilon=1.0, r_cut=2.0 ** (1 / 6))
Include the forces from the walls on the particles in the equations of motion:
[9]:
simulation.operations.integrator.forces.append(wall_lj)
Run the simulation#
[10]:
simulation.run(50_000)
[11]:
render(simulation.state.get_snapshot())
[11]:
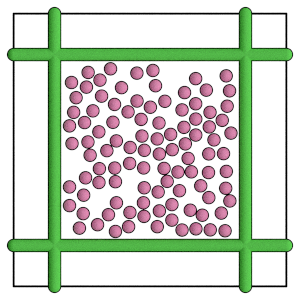
Conclusion#
This section demonstrates how to apply a force between wall geometries and particles during Molecular Dynamics simulations. The next section shows how to run the same simulation with HPMC.