md.dihedral¶
Overview
md.dihedral.harmonic |
Harmonic dihedral potential. |
md.dihedral.opls |
OPLS dihedral force |
md.dihedral.table |
Tabulated dihedral potential. |
Details
Dihedral potentials.
Dihedrals add forces between specified quadruplets of particles and are typically used to model rotation about chemical bonds.
By themselves, dihedrals that have been specified in an input file do nothing. Only when you specify an dihedral force (i.e. dihedral.harmonic), are forces actually calculated between the listed particles.
Important: There are multiple conventions pertaining to the dihedral angle (phi) in the literature. HOOMD utilizes the convention shown in the following figure, where vectors are defined from the central particles to the outer particles. These vectors correspond to a stretched state (phi=180 deg) when they are anti-parallel and a compact state (phi=0 deg) when they are parallel.
-
class
hoomd.md.dihedral.coeff¶ Defines dihedral coefficients.
The coefficients for all dihedral force are specified using this class. Coefficients are specified per dihedral type.
There are two ways to set the coefficients for a particular dihedral force. The first way is to save the dihedral force in a variable and call
set()directly. See below for an example of this.The second method is to build the
coeffclass first and then assign it to the dihedral force. There are some advantages to this method in that you could specify a complicated set of dihedral force coefficients in a separate python file and import it into your job script.Examples:
my_coeffs = dihedral.coeff(); my_dihedral_force.dihedral_coeff.set('polymer', k=330.0, r=0.84) my_dihedral_force.dihedral_coeff.set('backbone', k=330.0, r=0.84)
-
set(type, **coeffs)¶ Sets parameters for dihedral types.
Parameters: - type (str) – Type of dihedral, or list of types
- coeffs – Named coefficients (see below for examples)
Calling
set()results in one or more parameters being set for a dihedral type. Types are identified by name, and parameters are also added by name. Which parameters you need to specify depends on the dihedral force you are setting these coefficients for, see the corresponding documentation.All possible dihedral types as defined in the simulation box must be specified before executing run(). You will receive an error if you fail to do so. It is not an error, however, to specify coefficients for dihedral types that do not exist in the simulation. This can be useful in defining a force field for many different types of dihedrals even when some simulations only include a subset.
To set the same coefficients between many particle types, provide a list of type names instead of a single one. All types in the list will be set to the same parameters.
Examples:
my_dihedral_force.dihedral_coeff.set('polymer', k=330.0, r0=0.84) my_dihedral_force.dihedral_coeff.set('backbone', k=1000.0, r0=1.0) my_dihedral_force.dihedral_coeff.set(['dihedralA','dihedralB'], k=100, r0=0.0)
Note
Single parameters can be updated. If both
kandr0have already been set for a particle type, then executingcoeff.set('polymer', r0=1.0)will update the value ofr0and leave the other parameters as they were previously set.
-
-
class
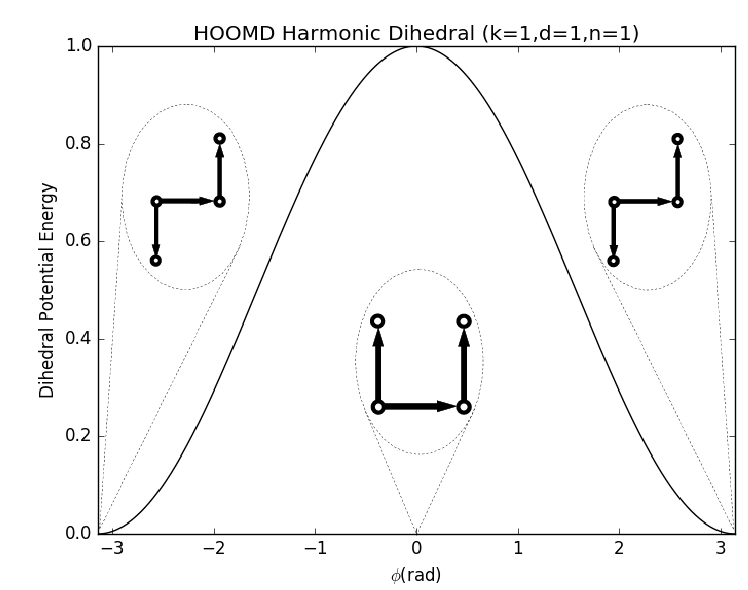
hoomd.md.dihedral.harmonic¶ Harmonic dihedral potential.
harmonicspecifies a harmonic dihedral potential energy between every defined dihedral quadruplet of particles in the simulation:\[V(r) = \frac{1}{2}k \left( 1 + d \cos\left(n * \phi(r) - \phi_0 \right) \right)\]where \(\phi\) is angle between two sides of the dihedral.
Coefficients:
- \(k\) - strength of force (in energy units)
- \(d\) - sign factor (unitless)
- \(n\) - angle scaling factor (unitless)
- \(\phi_0\) - phase shift
phi_0(in radians) - optional: defaults to 0.0
Coefficients \(k\), \(d\), \(n\) must be set for each type of dihedral in the simulation using
dihedral_coeff.set().Examples:
harmonic.dihedral_coeff.set('phi-ang', k=30.0, d=-1, n=3) harmonic.dihedral_coeff.set('psi-ang', k=100.0, d=1, n=4, phi_0=math.pi/2)
-
disable(log=False)¶ Disable the force.
Parameters: log (bool) – Set to True if you plan to continue logging the potential energy associated with this force. Examples:
force.disable() force.disable(log=True)
Executing the disable command will remove the force from the simulation. Any
hoomd.run()command executed after disabling a force will not calculate or use the force during the simulation. A disabled force can be re-enabled withenable().By setting log to True, the values of the force can be logged even though the forces are not applied in the simulation. For forces that use cutoff radii, setting log=True will cause the correct r_cut values to be used throughout the simulation, and therefore possibly drive the neighbor list size larger than it otherwise would be. If log is left False, the potential energy associated with this force will not be available for logging.
-
get_energy(group)¶ Get the energy of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the energy for.Returns: The last computed energy for the members in the group. Examples:
g = group.all() energy = force.get_energy(g)
-
get_net_force(group)¶ Get the force of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the force for.Returns: The last computed force for the members in the group. Examples
g = group.all() force = force.get_net_force(g)
-
get_net_virial(group)¶ Get the virial of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the virial for.Returns: The last computed virial for the members in the group. Examples
g = group.all() virial = force.get_net_virial(g)
-
class
hoomd.md.dihedral.opls¶ OPLS dihedral force
oplsspecifies an OPLS-style dihedral potential energy between every defined dihedral.\[V(r) = \frac{1}{2}k_1 \left( 1 + \cos\left(\phi \right) \right) + \frac{1}{2}k_2 \left( 1 - \cos\left(2 \phi \right) \right) + \frac{1}{2}k_3 \left( 1 + \cos\left(3 \phi \right) \right) + \frac{1}{2}k_4 \left( 1 - \cos\left(4 \phi \right) \right)\]where \(\phi\) is the angle between two sides of the dihedral and \(k_n\) are the force coefficients in the Fourier series (in energy units).
\(k_1\), \(k_2\), \(k_3\), and \(k_4\) must be set for each type of dihedral in the simulation using
dihedral_coeff.set().Example:
opls_di.dihedral_coeff.set('dihedral1', k1=30.0, k2=15.5, k3=2.2, k4=23.8)
-
disable(log=False)¶ Disable the force.
Parameters: log (bool) – Set to True if you plan to continue logging the potential energy associated with this force. Examples:
force.disable() force.disable(log=True)
Executing the disable command will remove the force from the simulation. Any
hoomd.run()command executed after disabling a force will not calculate or use the force during the simulation. A disabled force can be re-enabled withenable().By setting log to True, the values of the force can be logged even though the forces are not applied in the simulation. For forces that use cutoff radii, setting log=True will cause the correct r_cut values to be used throughout the simulation, and therefore possibly drive the neighbor list size larger than it otherwise would be. If log is left False, the potential energy associated with this force will not be available for logging.
-
get_energy(group)¶ Get the energy of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the energy for.Returns: The last computed energy for the members in the group. Examples:
g = group.all() energy = force.get_energy(g)
-
get_net_force(group)¶ Get the force of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the force for.Returns: The last computed force for the members in the group. Examples
g = group.all() force = force.get_net_force(g)
-
get_net_virial(group)¶ Get the virial of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the virial for.Returns: The last computed virial for the members in the group. Examples
g = group.all() virial = force.get_net_virial(g)
-
-
class
hoomd.md.dihedral.table(width, name=None)¶ Tabulated dihedral potential.
Parameters: tablespecifies that a tabulated dihedral force should be applied to every define dihedral.\(T_{\mathrm{user}}(\theta)\) and \(V_{\mathrm{user}}(\theta)\) are evaluated on width grid points between \(-\pi\) and \(\pi\). Values are interpolated linearly between grid points. For correctness, you must specify the derivative of the potential with respect to the dihedral angle, defined by: \(T = -\frac{\partial V}{\partial \theta}\).
Parameters:
- \(T_{\mathrm{user}}(\theta)\) and \(V_{\mathrm{user}} (\theta)\) - evaluated by
func(see example) - coefficients passed to func - coeff (see example)
Set table from a given function
When you have a functional form for V and T, you can enter that directly into python.
tablewill evaluate the given function over width points between \(-\pi\) and \(\pi\) and use the resulting values in the table:def harmonic(theta, kappa, theta0): V = 0.5 * kappa * (theta-theta0)**2; F = -kappa*(theta-theta0); return (V, F) dtable = dihedral.table(width=1000) dtable.dihedral_coeff.set('dihedral1', func=harmonic, coeff=dict(kappa=330, theta_0=0.0)) dtable.dihedral_coeff.set('dihedral2', func=harmonic,coeff=dict(kappa=30, theta_0=1.0))
Set a table from a file
When you have no function for for V or T, or you otherwise have the data listed in a file, dihedral.table can use the given values directly. You must first specify the number of rows in your tables when initializing
table. Then useset_from_file()to read the file.dtable = dihedral.table(width=1000) dtable.set_from_file(‘polymer’, ‘dihedral.dat’)-
disable(log=False)¶ Disable the force.
Parameters: log (bool) – Set to True if you plan to continue logging the potential energy associated with this force. Examples:
force.disable() force.disable(log=True)
Executing the disable command will remove the force from the simulation. Any
hoomd.run()command executed after disabling a force will not calculate or use the force during the simulation. A disabled force can be re-enabled withenable().By setting log to True, the values of the force can be logged even though the forces are not applied in the simulation. For forces that use cutoff radii, setting log=True will cause the correct r_cut values to be used throughout the simulation, and therefore possibly drive the neighbor list size larger than it otherwise would be. If log is left False, the potential energy associated with this force will not be available for logging.
-
get_energy(group)¶ Get the energy of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the energy for.Returns: The last computed energy for the members in the group. Examples:
g = group.all() energy = force.get_energy(g)
-
get_net_force(group)¶ Get the force of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the force for.Returns: The last computed force for the members in the group. Examples
g = group.all() force = force.get_net_force(g)
-
get_net_virial(group)¶ Get the virial of a particle group.
Parameters: group ( hoomd.group) – The particle group to query the virial for.Returns: The last computed virial for the members in the group. Examples
g = group.all() virial = force.get_net_virial(g)
-
set_from_file(dihedralname, filename)¶ Set a dihedral pair interaction from a file.
Parameters: The provided file specifies V and F at equally spaced theta values.
Example:
#t V T -3.141592653589793 2.0 -3.0 -1.5707963267948966 3.0 -4.0 0.0 2.0 -3.0 1.5707963267948966 3.0 -4.0 3.141592653589793 2.0 -3.0
Note
The theta values are not used by the code. It is assumed that a table that has N rows will start at \(-\pi\), end at \(\pi\) and that \(\delta \theta = 2\pi/(N-1)\). The table is read directly into the grid points used to evaluate \(T_{\mathrm{user}}(\theta)\) and \(V_{\mathrm{user}}(\theta)\).
- \(T_{\mathrm{user}}(\theta)\) and \(V_{\mathrm{user}} (\theta)\) - evaluated by